| Reference: | Martín-Hernández I, Chacón P, and
López-Blanco JR (2026).
AbRaCD: Fast sampling-based prediction of antibody CDR loops. (Submitted)
|
| *Freely accessible to all users, including commercial users, with no login or registration required. | |
Gallery

Help
AbRaCD is a sampling-based web server for ab initio prediction of antibody complementarity-determining region (CDR) loops, designed to capture antibody-specific conformational features. Building on RCD+, it introduces CDR-dependent Ramachandran restrictions, explicit treatment of the H3 kink, an improved backbone-aware KORP energy filter, and a final full-atom refinement stage. Our fast, automated, easy-to-use platform for CDR loop modeling achieves competitive performance compared with current state-of-the-art approaches and provides multiple ranked conformations.
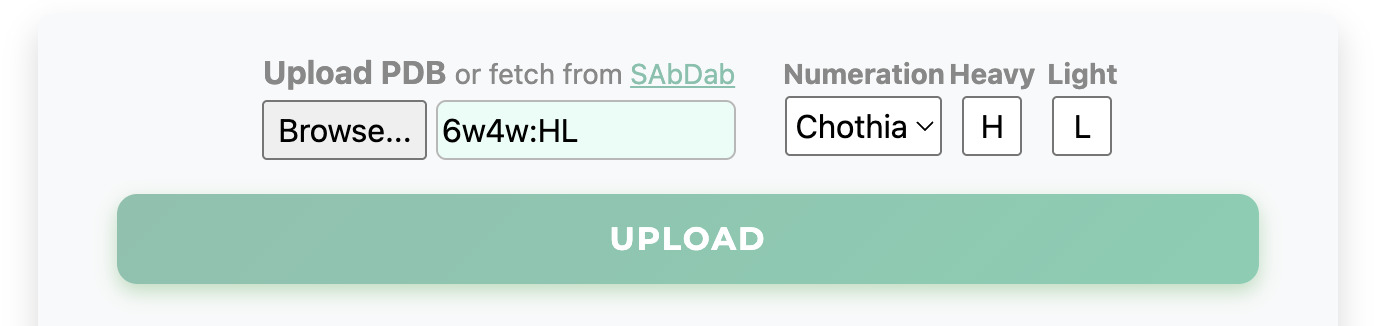
Users can upload an antibody PDB structure numbered according to the Chothia scheme, or fetch
structures
directly from the SAbDab database by entering the PDB ID.
AbRaCD follows the same Chothia* numeration convention as SAbDab. We strongly recommend tools like AbNum
[Abhinandan & Martin, 2008] to generate a Chothia numbered input PDB.
Alternatively, select "Raw" numeration to upload a sequentially numbered PDB file and manually provide
the correct anchor residue indices.
Heavy (H) and Light (L) chain identifiers are required. If the fields are left empty, the server uses H and L by default. Users must ensure that the specified chain IDs are present in the input PDB; otherwise, an error will be returned.
Upon upload, AbRaCD automatically parses the structure and, only for Chothia numeration, identifies all CDR loops and extracts the corresponding anchor residues and chain identifiers.
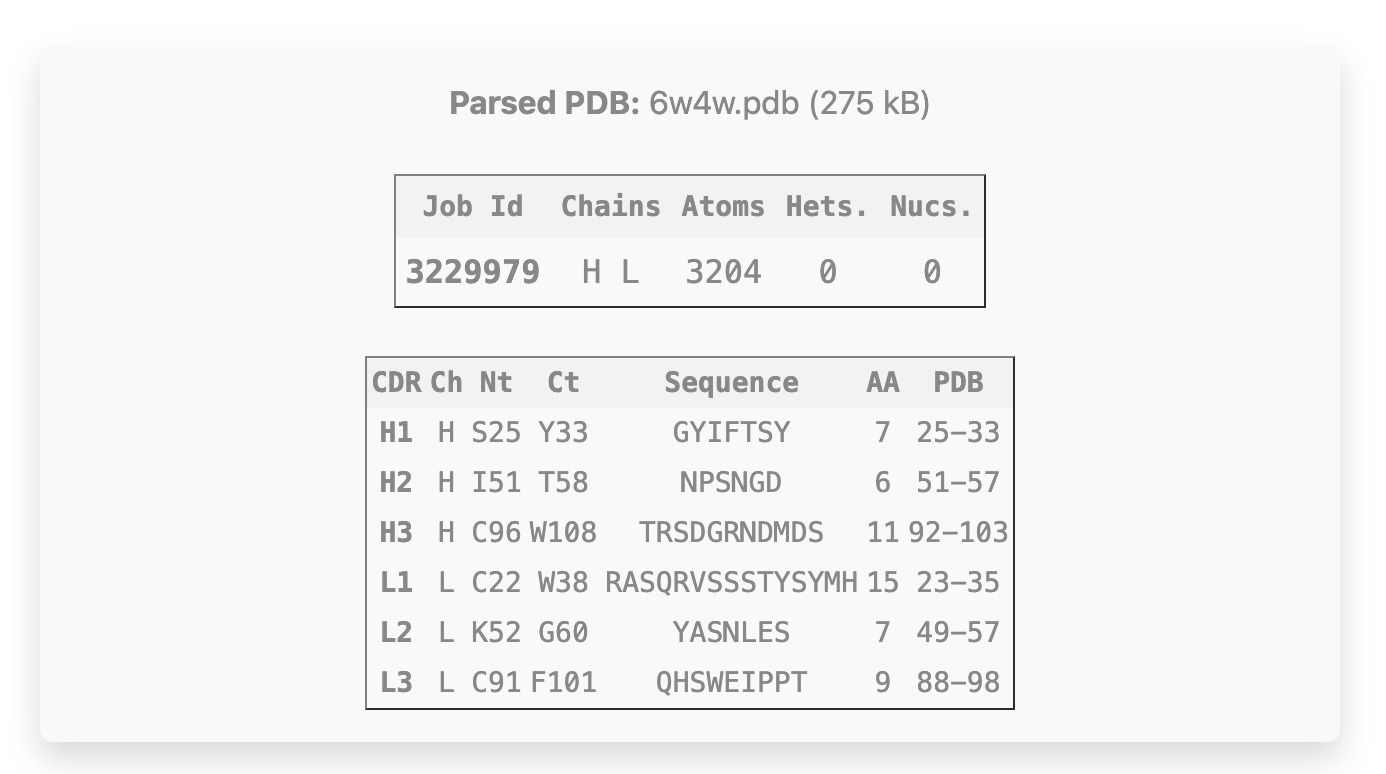
The table shows the N-terminal (Nt) and C-terminal (Ct) anchor residues of each CDR loop along with
their sequences (if present in PDB) and loop lengths (AA).
The anchor indices in columns "Nt" and "Ct" correspond to the parsed PDB (sequentially renumbered).
The
input PDB anchor indices are shown in "PDB" column.
Note that anchor residues are the fixed residues flanking the modeled region, and their presence in
the PDB is mandatory.
If present in PDB, the CDR loop sequences (between anchors) and lengths are shown in Sequence and AA columns, respectively. Note that the loop coordinates are never used for prediction and serve only for final RMSD analysis and validation purposes. If the entire sequence between anchors is missing, the CDR will be highlighted in yellow. In this case, the input sequence should be provided in the next step.
*CDR-L1:L24-34; CDR-L2:L50-56; CDR-L3:L89-97; CDR-H1:H26-32; CDR-H2:H52-56; CDR-H3:H95-102.
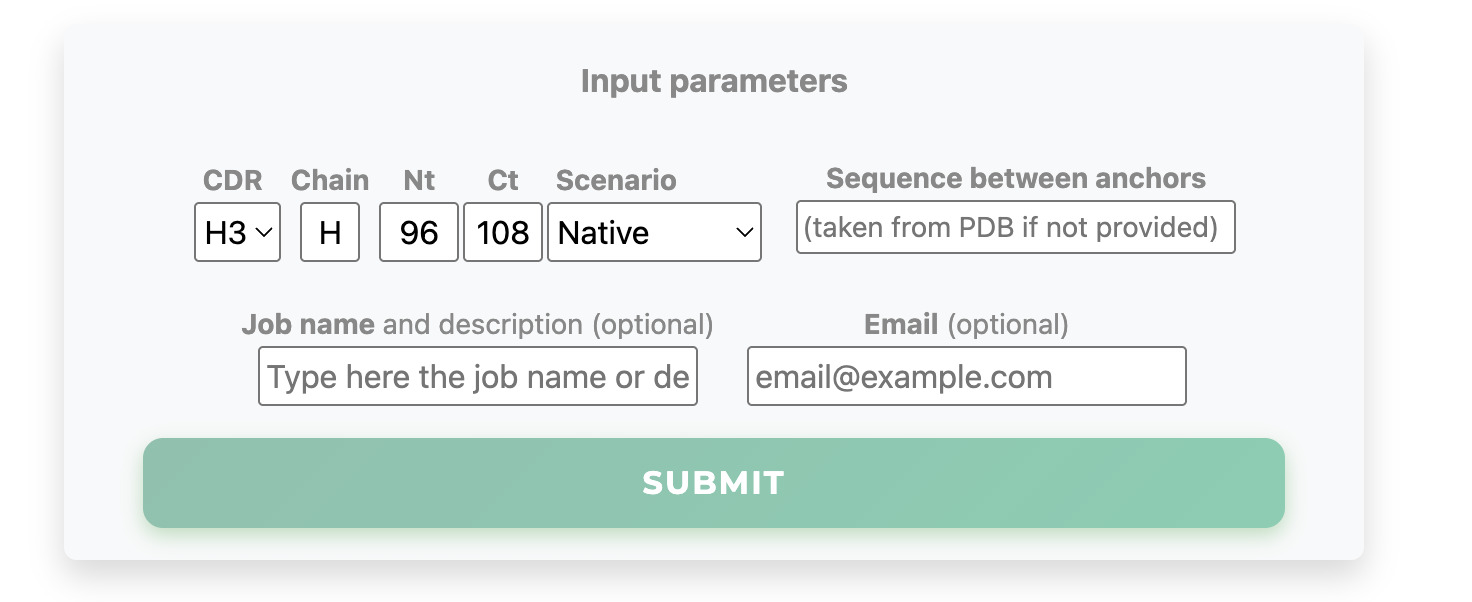
The user selects the CDR loop to be modeled and may optionally provide any loop sequence (20 residues maximum). If you want to compare predictions with the native structure present in the PDB, do not provide any sequence.
Three prediction scenarios are available: Native (loop modeling only), Modeling (loop modeling with side-chain optimization of the surrounding environment), and Relax+Mod (initial full-structure relaxation followed by Modeling).
Optionally, the job can be named, and an email address (recommended) can be provided to access the results at any time for up to two months.
Finally, select your desired CDR loop and click Submit.
Jobs are queued immediately and marked as pending (PD), running (R), or finished.
Typical runtimes range from 1-2 minutes for short loops (6-8 residues) up
to 20-30 minutes for long loops (≥12 residues). Up to three jobs may be queued concurrently.
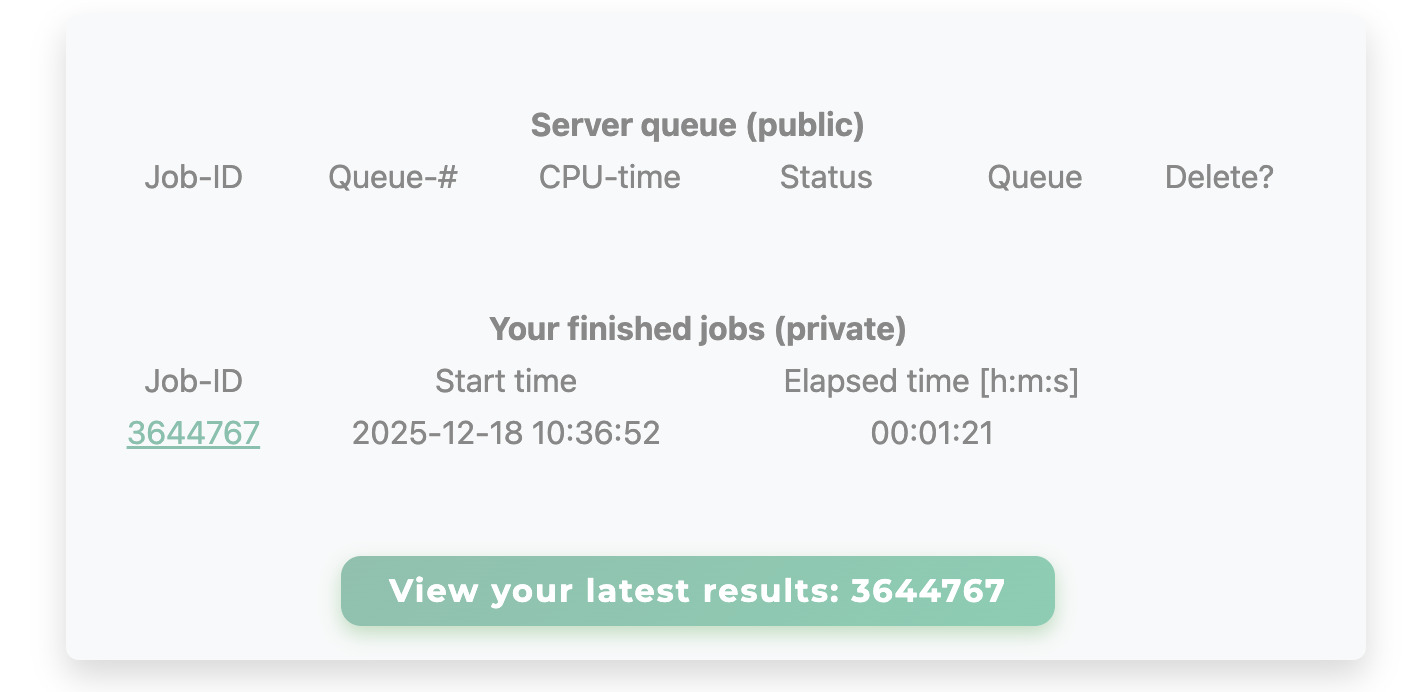
Jobs are assigned a permanent results URL at submission time, which reports job status (queued, running, finished) and allows bookmarking. Results are kept available for at least two months.
AbRaCD generates multiple candidate loop conformations ranked by a knowledge-based energy function. The 10–20 best refined loops are visualized interactively in the browser using JSmol together with the input antibody structure.
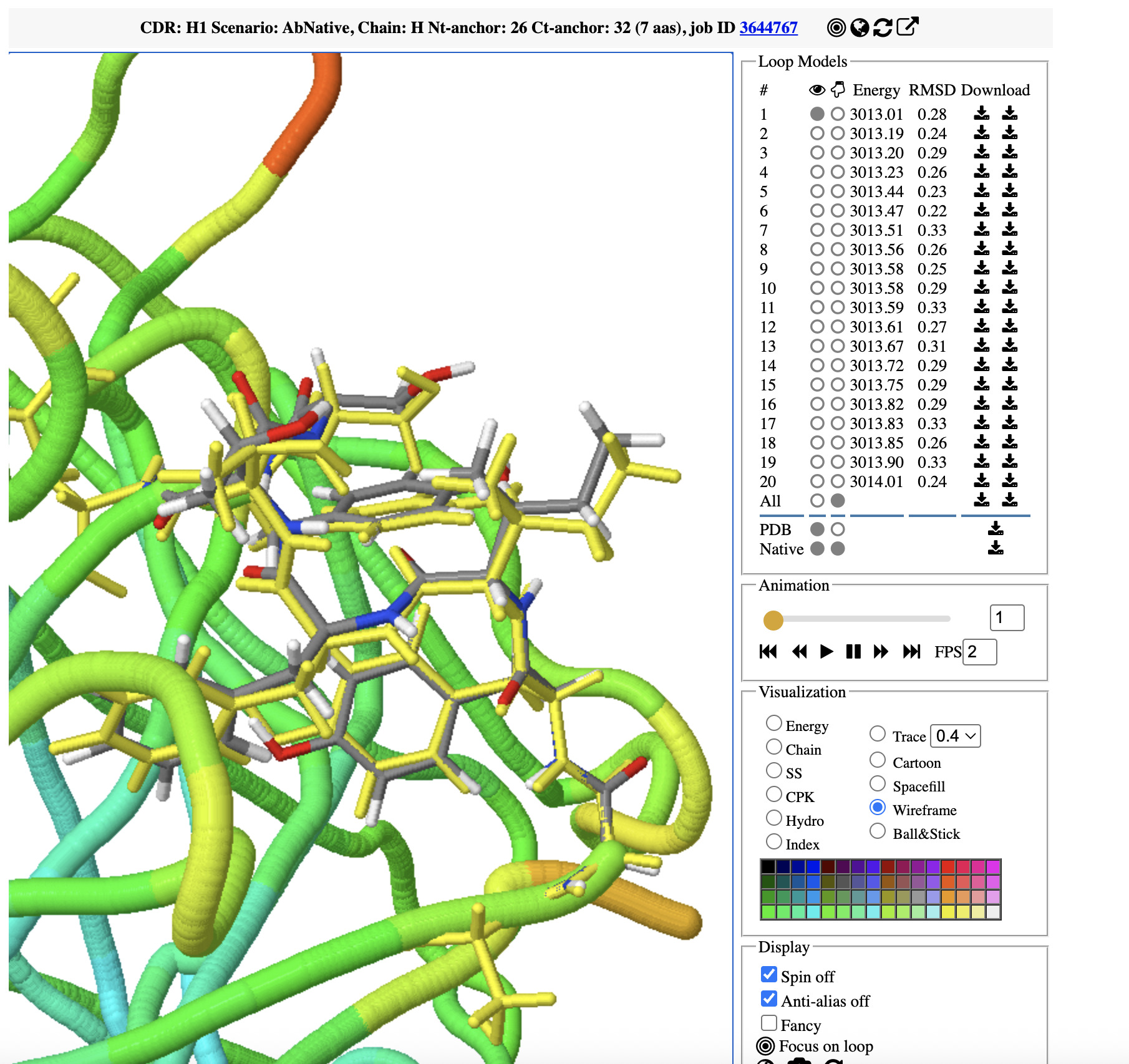
Use mouse controls to explore the generated loops and customize their molecular representation and colors. For example, drag to rotate, Shift + double-left-click (or Shift + right-click) to translate, or use the palette to recolor selected loop(s).
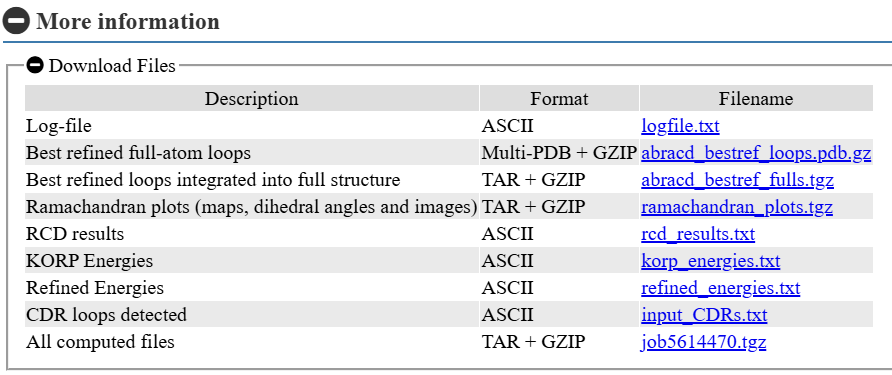
Predicted loops can be downloaded either as isolated loop PDB files or integrated into the full antibody structure, and all job-related files are available for download either individually or together as a single Tar+GZip archive.
Additional graphical output includes antibody-specific Ramachandran maps with predicted φ/ψ angles overlaid, as well as diagnostic energy-versus-RMSD plots before and after full-atom refinement.
You can retrieve results at any time by entering the job ID number here:

Handling multiple chains
Antibody PDB files often include additional chains beyond the Heavy (H) and Light (L) chains. These additional
chains may correspond to antigens, multiple Fv fragments, or other molecules.
For example, the PDB entry 1ahw has six chains (A, B, C, D, E, and F)
forming two Fv fragments: B/A and E/D (Heavy/Light).
You can find the correspondence between chains and Fv(s) by checking the 1ahw
SAbDab
page.
In AbRaCD, you can select exactly which chains to model by simply entering the required Heavy and
Light chain IDs in the corresponding input fields:
Fv B/A: Fetch 1ahw with “1ahw:BA” (or upload the PDB file) and set "H chain" to "B" and "L chain" to "A".
Fv E/D: Fetch 1ahw with “1ahw:ED” (or upload the same PDB file) and
set "H chain" to "E" and "L chain" to "D".
L3 loops frequently contain cis-proline residues
For example, in L3 loops that are 9 residues long, the 7th residue is a cis-proline in most cases.
To obtain optimal results, cis-prolines must be explicitly indicated in the input sequence using a
lowercase "p":
cis-Proline L3: Fetch 2hwz with “2hwz:HL” and input the sequence:
"CFQGSGYpFTF".